
04478
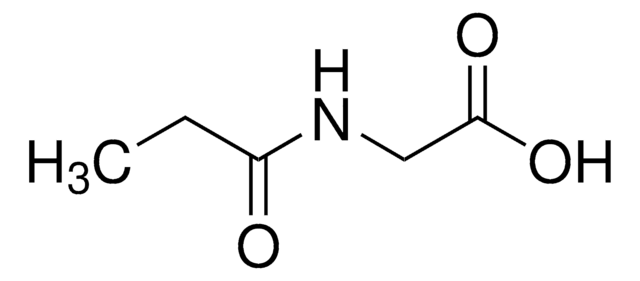
N-(2-Methylbutyryl)glycine
analytical standard
Synonym(s):
2-Methylbutyryl glycine, N-(2-Methyl-1-oxobutyl)glycine
About This Item
Recommended Products
grade
analytical standard
Quality Level
assay
≥98.0% (HPLC)
shelf life
limited shelf life, expiry date on the label
application(s)
clinical testing
format
neat
storage temp.
2-8°C
InChI
1S/C7H13NO3/c1-3-5(2)7(11)8-4-6(9)10/h5H,3-4H2,1-2H3,(H,8,11)(H,9,10)
InChI key
HOACIBQKYRHBOW-UHFFFAOYSA-N
Related Categories
Biochem/physiol Actions

signalword
Warning
hcodes
Hazard Classifications
Eye Irrit. 2 - Skin Irrit. 2
wgk_germany
WGK 3
flash_point_f
Not applicable
flash_point_c
Not applicable
Choose from one of the most recent versions:
Certificates of Analysis (COA)
Don't see the Right Version?
If you require a particular version, you can look up a specific certificate by the Lot or Batch number.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service